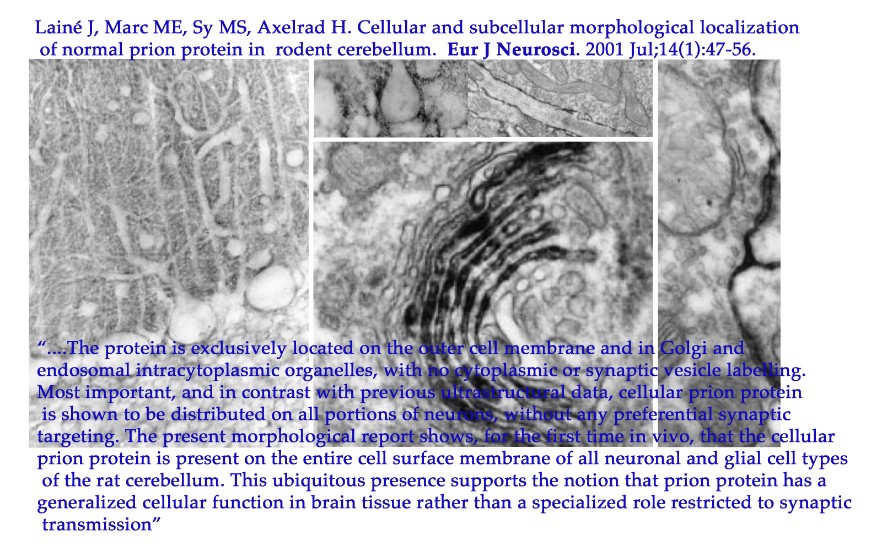
 |
||||
LINK Transmissible spongiform encephalopathies (TSEs or prion diseases) are a rare group of invariably fatal neurodegenerative disorders that affect humans and other mammals. TSEs are protein misfolding diseases that involve the accumulation of an abnormally aggregated form of the normal host prion protein. They are unique among protein misfolding disorders in that they are transmissible and have different strains of infectious agent that are associated with unique phenotypes in vivo. |
LINK Prion diseases are transmissible spongiform encephalopathies (TSEs), attributed to conformational conversion of the cellular prion protein (PrPC) into an abnormal conformer that accumulates in the brain. Understanding the pathogenesis of TSEs requires the identification of functional properties of PrPC. Here we examine the physiological functions of PrPC at the systemic, cellular, and molecular level |
LINK Prion diseases are caused by conversion of a normal cell-surface glycoprotein (PrPC) into a conformationally altered isoform (PrPSc) that is infectious in the absence of nucleic acid. Although a great deal has been learned about PrPSc and its role in prion propagation, much less is known about the physiological function of PrPC. In this review, we will summarize some of the major proposed functions for PrPC, including protection against apoptotic and oxidative stress, cellular uptake or binding of copper ions, transmembrane signaling, formation and maintenance of synapses, and adhesion to the extracellular matrix. |
||